CD-HIT main paradigm. Source: Chen, Q. et al. (2018) Journal of Data and Information Quality.
Turn A Collection of Protein Structures into a Cluster of Non-Redundant Sequences with pdb2fasta, SeqKit and CD-HIT
Posted: 2026-02-07
Creating non-redundant (NR) databases of sequences is essential to analyzing and evaluating many methods in bioinformatics. In this article, the process of creating such a list of sequences is shown with pdb2fasta and CD-HIT programs (with the optional use of seqkit to filter sequences), building a list of representative proteins for evaluating structure search methods (commonly evaluated with a set of proteins that share relatively low sequence similarity - much like one would balance classes for a more fair evaluation of classification methods).
There are several programs and APIs that can convert a collection of protein 3D structures (often in PDB format) into their amino acid sequences (e.g., the pdb2fasta CLI, BioPython library, etc.), which simply parse the PDB files for amino acid order. Then, one can take the collection of sequences (often represented in fasta format) and cluster them with a certain sequence similarity cutoff to get less redundancy. This is useful for reducing a collection of sequences that may be too large and/or contain too many similar sequences or duplicates for robust downstream analysis.
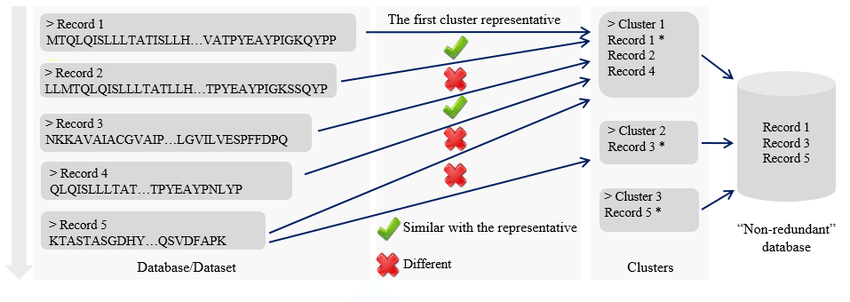
With CD-HIT this reduction is done by aligning sequences using some clever optimizations and filters for speeding things up like an index table and short word filter (more in their user's guide and paper - see References below). The clustering technique avoids all-by-all pairwise clustering (like one would do with BLAST), a potentially very computationally expensive procedure, by using an incremental approach with word-based heuristics. Instead of the typical k-mer (substring of length k) hash table speed-up (k-mer to an index value) that BLASTN and BLASTP uses, CD-HIT uses a faster index table scheme that maps unique indices to all possible unique k-mers, holding it in memory for computational speed. In CD-HIT, sequences are sorted from longest to shortest, processing them sequentially. At initialization that longest sequence is considered the representative of it's cluster (cluster of 1). Subsequent sequences are all compared to the previous representatives of clusters and assigned to the first (in fast mode) or best (in accurate mode) cluster based on sequence similarity and a sequence similarity cutoff determining how related the clusters will be, creating new clusters as needed.
The following is an example of extracting sequences from protein structure files and then running CD-HIT to find a representative list with less than 40% sequence similarity.
- Download 3D structure files. Here, the SCOPe 2.08 protein structure database was used (which can be downloaded from https://zenodo.org/records/5829561). The creators of SCOPe do provide sequences, but for demonstrative purposes here, the sequences are extracted from the structures.
- Leverage the
pdb2fastacommand line tool to extract the sequences from a directory of PDB files (see Appendix for a bash script example withpdb2fasta).- Download the
pdb2fastaexecutable for Linux withwget https://aideepmed.com/pdb2fasta/pdb2fasta(and optionally place in/usr/local/bin). Note, if this tool is no longer available, try BioPython (Python snippet below in Appendix). - Make
pdb2fastaexecutablechmod +x pdb2fasta. - Run this in a loop in bash over a directory of PDB files (see Appendix) to create a fasta-style file of sequences.
- Download the
- [Optional] Filter the fasta sequence file with the
seqkit(links below) tool to find sequences between certain legnth cutoffs (here, 20-1000 amino acids), where-mis the lower limit of sequence length (inclusive),-Mis the upper limit of sequence length (inclusive), and-ois the output file name.- Download the
seqkitexecutable for Linux (it is available for Windows and macOS as well) withwget https://github.com/shenwei356/seqkit/releases/download/v2.11.0/seqkit_linux_amd64.tar.gz, extract withtar -zxvf seqkit_linux_amd64.tar.gz(and optionally place in/usr/local/bin) seqkit seq -m 20 -M 1000 -o pdbstyle-2.08_20to1000aa.fasta pdbstyle-2.08.fasta
- Download the
- Leverage CD-HIT to cluster and find representative sequences, where
-iis the input,-ois the output,-nis the word length,-cis the sequence similarity cutoff,-lis the min sequence cutoff (this value or less),-M 0for unlimited memory limit, and-T 0for max number of threads (using all CPUs). See the user's guide (https://github.com/weizhongli/cdhit/wiki/3.-User's-Guide) for more info.cd-hit -i pdbstyle-2.08_20to1000aa.fasta -o pdbstyle-2.08_40seqsim.lst -n 2 -c 0.4 -l 19 -M 0 -T 0- Note: a limitation of a short word length filter (here it is 2) is that it can not be used below certain clustering thresholds (here, 0.4 for 40%).
This results is several fasta-style files - pdbstyle-2.08.fasta - pdbstyle-2.08_20to1000aa.fasta - pdbstyle-2.08_40seqsim.lst
As well as a CD-HIT list of proteins that belong to clusters. - pdbstyle-2.08_40seqsim.lst.clstr
Interestingly, the CD-HIT project has great tools for NGS data processing like removing read duplicates, finding pairs of overlapping reads or joining pair-end reads etc.
Also, seqkit is a hugely versatile library for bioinformatics (format conversions, search, BAM processing, sampling, stats, etc.). Check the Additional References for links.
References
pdb2fasta
SeqKit
- Shen, W., Sipos, B. & Zhao, L. (2024) "SeqKit2: A Swiss Army Knife for Sequence and Alignment Processing." iMeta e191. doi:10.1002/imt2.191
- SeqKit - a cross-platform and ultrafast toolkit for FASTA/Q file manipulation
CD-HIT
- Li, W. & Godzik, A. (2006) "Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences." Bioinformatics 22:1658-1659. PubMed
- Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. (2012) "CD-HIT: accelerated for clustering the next generation sequencing data." Bioinformatics 28:3150-3152. doi:10.1093/bioinformatics/bts565
- CD-HIT GitHub
- CD-HIT clustering algorithm
- CD-HIT user's guide
BLAST (for comparison)
- Altschul, S.F., Gish, W., Miller, W., Myers, E.W., Lipman, D.J. (1990) "Basic local alignment search tool." J. Mol. Biol. 215:403-410. PubMed
- Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., Madden T.L. (2008) "BLAST+: architecture and applications." BMC Bioinformatics 10:421. PubMed
- BLAST - NCBI
Appendix
Example bash script to run pdb2fasta over a directory of PDB structure files (which is called pdbstyle-2.08, here):
#!/bin/bash
# Define the directory you want to start from
start_dir="pdbstyle-2.08"
echo "Starting recursive loop using find in $start_dir"
# Use find to list files and loop through the output
# -type f ensures only regular files are returned
# -print0 and read -d '' safely handle filenames with spaces or special characters
find "$start_dir" -type f -print0 | while IFS= read -r -d '' file; do
echo "Processing file: $file"
./pdb2fasta "$file" >> "pdbstyle-2.08.fasta"
done
Get a protein sequence from a PDB file with BioPython library example:
from Bio.PDB.PDBParser import PDBParser
from Bio.PDB.Polypeptide import PPBuilder
def get_sequence(pdb_path):
"""Get a protein sequence from a PDB file"""
seq = ''
try:
p = PDBParser(PERMISSIVE=0)
structure = p.get_structure('xyz', pdb_path)
ppb = PPBuilder()
for pp in ppb.build_peptides(structure):
seq += pp.get_sequence()
except Exception:
seq = ''
return seq